Alpha-Synuclein (α-syn)
Original Editor - Lucinda hampton
Top Contributors - Lucinda hampton and Uchechukwu Chukwuemeka
Introduction[edit | edit source]

Alpha-synuclein (α-syn) is a small protein encoded by the SNCA gene (the gene that provides instructions for α-syn). It is amply expressed in the nervous system and exists in a natively unfolded state under normal physiological conditions (however its conformation changes depending on its environment and interactions with binding partners).[1]
Where Is α-Syn[edit | edit source]
α-syn is abundant in the brain, and smaller amounts are found in the heart, muscles, and other tissues. In the brain, α-syn is found mainly at the tips of neurons in the specialized structures known as presynaptic terminals. Presynaptic terminals release neurotransmitters (chemical messages), from synaptic vesicles. In a healthy nervous system, α-syn is involved in transmission between neurons, helping signals move throughout the nervous system and within the brain. This allows for coordinated movement, proper control of bodily functions, and overall normal brain function.[2]

Misfolding[edit | edit source]

For our body to function correctly, proteins are folded neatly into a shape that allows them to do their jobs . Proteins that are structured incorrectly usually are flagged and then fixed or thrown away. This is a complicated job involving enzymes, chaperones, and small organelles, all monitoring the protein shape. This network helps prevent the formation of sticky piles of protein (known as aggregates) that cause disarray to our cells.
However, the presence of genetic mutations or simply aging can make the protein surveillance system less effective, and this allows proteins (for example α-syn) to turn nasty, folding into an incorrect shape that cannot be fixed. If the neuron is unable to fix or dispose of the wrong shape, a whole a cascade of protein-misfolding occurs results in a sticky pile of α-syn. This causes havoc to the the normal neuronal function, creating debris that the neuron may not be able to clean up.
Many conditions (see synucleinopathies) can promote this transition from soluble, functional α-syn into insoluble aggregates, and once this process begins, it proceeds in a feed-forward manner in aggregates form and seed, recruiting soluble α-syn into more aggregates, a process that once escalated is largely irreversible.
While α-syn oligomers and aggregates have not been proven to be directly toxic, they are consistently associated with toxicity. Within Parkinson's disease and other synucleinopathies, α-syn transforms from a soluble, functional protein to aggregated protein (i.e., LBs) that becomes associated with pathogenic results.[3][2]

Lewy Bodies[edit | edit source]
The aggregation formation involves the binding between α-syn and other protein complexes (examples being tau protein and β-amyloid) and is termed “cross-seeding,”. This mechanism is largely responsible for the formation of Lewy bodies (LB). LB are abnormal neuronal intracytoplasmic inclusions which are composed of > 70 proteins, the core being chiefly α-syn fibrillar aggregates.[4]

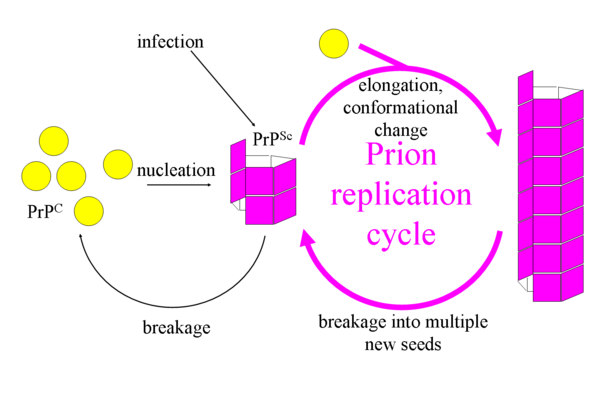
Prion-Like Characteristics of α-Syn[edit | edit source]
A prion is a type of protein that can cause disease in animals and humans by triggering normally healthy proteins in the brain to fold abnormally. Through this process, it generates a great quantity of misfolded proteins, which are pathological for the carrier (patient). A core characteristic of prion proteins is their inter-organism transmissibility, which makes prions infectious agent (with a high morbidity).
- To date, no evidence has been found supporting α-syn aggregation formation being transmissible from one organism to another in Parkinson's Disease. There is however evidence of α-syn propagation from the gut to the brain.
- MSA (another synucleinopathy) is classified as a prion disease due to the evidence that inoculation of samples from brains of MSA patients into mice brains causes aggregation and spreading of α-syn.
- Regarding Lewy Body Dementia, more information is needed to know if it has a transmission mechanism like prions.[4]

References[edit | edit source]
- ↑ Medline plus SNCA gene Available from: https://medlineplus.gov/genetics/gene/snca/ (accessed 16.9.2022)
- ↑ 2.0 2.1 Miller KM, Mercado NM, Sortwell CE. Synucleinopathy-associated pathogenesis in Parkinson’s disease and the potential for brain-derived neurotrophic factor. npj Parkinson's Disease. 2021 Apr 12;7(1):1-9.
- ↑ Gain therapeutics What is alpha-synuclein and what is its role in neurodegenerative diseases? Available from:https://perspectives.gaintherapeutics.com/what-is-alpha-synuclein-and-its-role-in-neurodegenerative-diseases/ (accessed 16.9.2022)
- ↑ 4.0 4.1 Bernal-Conde LD, Ramos-Acevedo R, Reyes-Hernández MA, Balbuena-Olvera AJ, Morales-Moreno ID, Argüero-Sánchez R, et al. Alpha-synuclein physiology and pathology: A perspective on cellular structures and organelles. Frontiers in neuroscience. 2020 Jan 23;13:1399.

