Gaucher Disease
Original Editors - Sarah Ansburg from Bellarmine University's Pathophysiology of Complex Patient Problems project.
Top Contributors - Sarah Ansburg, Admin, Elaine Lonnemann, Kim Jackson, WikiSysop and Wendy Walker
Definition/Description[edit | edit source]
An autosomal recessive inherited genetic disorder of metabolism in

which a dangerous level of a fatty substance called glucocerebroside collects in the liver, spleen, bone marrow, lungs, and at times in the brain. Gaucher disease is caused by mutations in a gene called GBA. Changes in the GBA gene cause low levels of glucocerebrosidase.[1] An individual with thisdisorder inherits a mutated copy of the GBA gene from each of his or her parents. Signs and symptoms of this disease can vary broadly among individuals and types.[2]
Prevalence[edit | edit source]
Gaucher disease occurs in about 1 in 50,000 to 100,000 individuals in the overall general population. Type 1 is the most common form and is frequent among individuals who are of Ashkenazi Jewish ancestry. Type 1 is seen in 1 in 500 to 1000 people of this type of Jewish descent and roughly 1 in 14 Ashkenazi Jews is a carrier. Type 2 and 3 of this disease are not as common.[4]

Characteristics/Clinical Presentation[edit | edit source]
Signs and symptoms for this disorder vary greatly upon the type of the disorder and the individual themselves, but may include:[6]
- Fractures & bone pain [7]
- Easy to bruise
- Cognitive problems
- Enlarged spleen (splenomegaly)
- Enlarged liver (hepatomegaly)
- Skin Changes
- Heart valve impairments
- Seizures
- Lung Diseases
- Swelling at birth
- Fatigue

| Type |
Presentation |
| Type 1 |
Most common form of this condition and is named Non-neuronopathic Gaucher Disease due to the lack of involvement of the brain and spinal cord.[4] Symptoms at this stage can range from very mild to severe at times and can develop at any age. Chief signs and symptoms may include enlargement of the spleen and liver (hepatosplenomegaly), a decrease in blood platelets causing, bruising (thrombocytopenia), lung disease, decrease in number of red blood cells (anemia), and bone defects such as deep pain felt in the bone, arthritis and fractures.[4]
|
| Type 2 |
Classified as a neuropathic form of the disease due to its involvement of the brain and spinal cord (central nervous system).[4] At this stage, liver and spleen enlargement can be evident as early as 3 months of age. Individuals may have substantial brain damage, seizures, abnormal eye movements and usually die prior to the age of 2.[2] |
| Type 3 |
Type 3 is also categorized as a neuronopathic disorder because it also affects the central nervous system, but progresses at a slower rate than type 2. At this stage, spleen and liver magnification is unpredictable, and brain association including seizures steadily become evident.[2] Key signs and symptoms can include eye movement disorders, blood disorders, and skeletal abnormalities.[2] |
| Perinatal Lethal Form |
This category causes life-threatening problems beginning prior to birth or in infancy.[4]Elements of this category may include significant swelling caused by fluid accumulation before birth (hydrops fetalis), dry skin (ichthyosis) enlarged spleen and liver, distinct facial features; and severe neurological problems. Most infants with this form only survive for a few days after birth.[4] |
| Cardiovascular Type |
This form mainly affects the heart. It causes the heart valves to harden or calcify. Individuals with this form may also have eye abnormalities, swelling of the spleen and bone disease.[4] |

Associated Co-morbidities[edit | edit source]
Co-morbidities associated with Gaucher disease include the following:[9]
- Increased occurance of malignancy (multiple myeloma and other lymphoproliferative disorders)
- Impaired glucose tolerance
- Neurologic disorders (including parkinsonian syndrome and peripheral neuropathy)
Medications[edit | edit source]
- Enzyme Replacment Therapy (Cerezyme), (VPRIV), (Ceredase)
- Substrate Replacment Therapy (Miglustat (MIH glue stat)
- Bone Marrow Transplant
Diagnostic Tests/Lab Tests/Lab Values[edit | edit source]
- Diagnosis of Gaucher Disease is focused on lab testing and clustering of clinical signs/symptoms.
Gaucher disease is thought to be a possible diagnosis in individuals with the following:
- Enlarged liver/spleen (hepatosplenomegaly)
- Bone pain/abnormalities
- Tendency to bruise and bleed very easily from low platelets
- Possible signs of neurological involvement
- Alterations in red blood cell levels.[1]
Laboratory testing includes the following:
- Blood work and a blood test called an enzyme assay are used to determine the activity level of the enzyme
- Individuals diagnosed with Gaucher disease have extremely low levels of the glucocerebrosidase activity.
- Analysis of DNA. This analysis looks for the GBA gene and the four most frequent GBA mutations.[1]
- When the GBA gene mutation is known in a family, DNA testing should be completed and used to correctly recognize any carriers.[1]
Etiology/Causes[edit | edit source]
- Gaucher disease is caused by a mutated gene called GBA. It is inherited in an autosomal recessive way when a person has two copies of the genes that supply directions for constructing the enzyme,glucocerbrosidase.[1] For healthy people, both genes work correctly.[1] When one of the genes is not functioning appropriately the person becomes a carrier for the disorder.
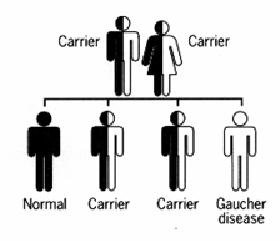

- Carriers do not have the disease because they have one functioning gene that creates adequate amounts of the enzyme for normal body function. When an individual inherits a mutated gene from each carrier parent, he or she then develops Gaucher disease.[1]
- Carrier parents have a 1 in 4 or 25% chance to conceive a child born with the disease. They have a 1 in 2, or 50 % chance to have a child who is a carrier, and a 1 in 4, or 25% chance to produce a child who is neither has the disease or is a carrier.[1]
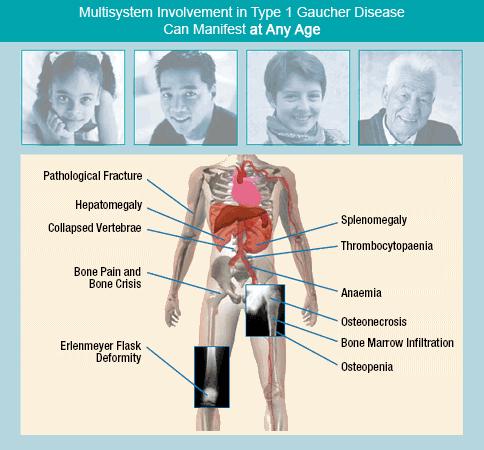
Systemic Involvement[edit | edit source]
Gastrointestinal[11]
- Splenomegaly (most common symptom in all three types)
- Hypersplenism contributing to pancytopenia
- Mechanical interference with bowel, kidney, diaphragm or circulation
- Hepatomegaly
Musculoskeletal[11]
- Replacement of bone marrow with Gaucher cells
- Bone pain
- Femoral head necrosis
- Pathologic fractures at the femoral neck and long bones
- Collapse of vertebral bodies with risk of thoracic kyphosis and spinal cord compression
- Bone infarction
Respiratory[11]
- Hypoxia due to pulmonary infiltrationand liver disease
- Restrictive lung disease
- Pulmonary function abnormalities
- Pulmonary hypertension
Cardiovascular[11]
- Restrictive pericarditis
- Myocardial infilitration
- Valve calicification
Dermatologic[11]
- Brown-yellow pigmentation of the skin
Neurological System [11]
- damage to the brain in types 2 and 3


Medical Management (current best evidence)[edit | edit source]
- Enzyme replacement therapy is utilized for individuals with types 1 and 3 of the disease. Enzyme therapy works by decreasing skeletal abnormalities, decreasing liver and spleen size/inflammation, and reversesother symptoms of the disorder, including abnormal blood counts. The treatment includes a modified form of the glucocerbrosidase by IV infusion every two weeks.[14]Enzyme therapy has no effects on the neurological aspects of the disorder.[14]
- The most effective dosing regimen of ERT is still debatable. Believers in of low dose regimens focus on the high costs of the enzyme. Others argue that high doses are necessary for the ideal effect to alleviate the severity of the disease, especially in children.[15]
- One strategy used is called top-down, with the dose being fairly high in the beginning and then slowly decreased to reach a maintenance level. [10]
- Another strategy used is called the step-up approach, in which the dose is low in the beginning and then built up if necessary, during the disease.[15]
- Both approaches have been compared in a recent study showing similar results for hematologic and visceral components, and a higher dose was better for improvement in surrogate factors including chitotriosidase and bone marrow involvement.[15]
- Some evidence shows that sudden cessation of therapy is not suggested due to a rebound phenomena occurance. A paper published by de Fost et al. shows that, in stable patients, the sudden reduction of the of the enzyme from weekly/biweekly infusions to a single monthly infusion, with a stable monthly dose may result in therapeutic failure in some patients.[15]
- A different study from Spain shows that after 2–3 years of initial biweekly enzyme replacement therapy treatment, infusion intervals were extended to 3 weeks with a 33% decline of the monthly dose. Within a few years, all of participants with the disease had to restart the original enzyme replacement therapy schedule because of a symptomatic setback of the disease.[15]

| Effects of Enzyme Replacement Therapy[15] | Description[15] |
| Symptomatic |
|
| Hematologic |
|
| Visceral |
|
| Skeletal |
|
| Children |
|
| Functional Health |
|
| Surrogate Parameters & Biomarkers |
|
U.S. Food and Drug Administration approved velaglucerase alfa for injection (VPRIV) for long-term enzyme replacement therapy for Type 1 Gaucher disease. VPRIV is an alternative to Cerezyme (imiglucerase), another enzyme replacement therapy which is currently in short supply.[16]
“The approval of VPRIV will provide a safe and effective alternative treatment for patients with Gaucher disease,” said Julie Beitz, M.D., director of the FDA’s Office of Drug Evaluation III. “Patients who previously received Cerezyme as an enzyme replacement therapy for their Type 1 Gaucher disease can be safely switched to VPRIV.”[16]
- Substrate Reduction Therapy (SRT) slows the build-up of unwanted fatty substance in Gaucher cells. Lysosomes then are able to try to catch up and eliminate the extra fatty substance.[17] SRT is taken orally by mouth every day in pill form.[17] It provides another option to enzyme replacement therapy.
- Miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) was introduced 6 years ago. This inhibits glucosylceramide synthase, which stops new synthesis of glucosylceramide in addition to the accumulated lipid.[15]
- There are currently less than ten clinical studies on miglustat published in the literature today. These current studies show that miglustat is useful in most patients with only mild disease at controlling the hematologic and visceral problems.[15]
- Miglustat is not tolerated as well as enzyme replacment therapy due to problems such as weight loss and diarrhea.

- Concerns about miglustat-induced cognitive problems have been resolved currently. The impact of miglustat, on neurological types of Gaucher disease is still under the investigation phase of development. In Europe, patients who cannot receive enzyme replacment therapy are able to receive miglustat as an alternative.[15]

- Bone marrow transplantation can help reverse the non-neurological effects of type 1, but has a high death rate due to defective donor matches.[2]
- Splenectomy
- There is no effective treatment for brain damage with types 2 and 3.[2]
Physical Therapy Management (current best evidence)[edit | edit source]
- There currently is no protocol or best treatment plans for gaucher disease, but exercise has been shown to be essential for children and adults that are diagnosed with Gaucher disease.
- Moderate exercise >3 days/week is recommended with a healthy diet for proper health and weight management
Reasoning for Exercise Include:[19]
- Counteract weight gain after enzyme replacement therapy due to better absorption in the GI tract & increase in appetite
- Exercise helps patients improve depression or sadness related to feeling wronged by the disease
- Help sufferers feel like they are in control of something about the disease and their life
- Help children feel like they belong and are normal like everyone else
Special Cautions to Exercise:[19]
- Talk with your physican before beginning exercise
- Those with enlarged spleens there is a possibility to bleed because of low platelet count, and all are advised to stay away from contact sports due to increased risk of pathological fracture
- Swimming is a good exercise for many patients due to gravity lessened movement. It strengthens muscles with minmized pressure on joint
- Those with hip/knee replacements should avoid exercises such as jogging and downhill skiing due to high impact.
Differential Diagnosis[edit | edit source]
The following may be associated with and/or differentially diagnosed for Gaucher disease:
- Saposin C Deficiency
- Lysosomal Storage Disorders
- Hepatosplenomegaly
- Niemann-Pick Disease
- Wolman Disease
- Mucopolysaccharidosis Type 1
- Mucopolysaccharidosis Type 2
- Legg-Calve Perthes Disease
- Hydrops Fetalis
- Mucopolysaccharidosis Type IV
- GM1 Gangliosidosis
- Metachromatic Leudodystrophy
- Myoclonic Seizures
- Thrombocytopenia
- Faber's Disease
- Multiple Myeloma[20]
- Hodgkin Disease[20]
- Lymphomas[20]
- Acute lymphocytic leukemia[7]
- Acute myelogenous leukemia[7]
Case Reports/ Case Studies[edit | edit source]
- Case Report-Gaucher Disease Type 2 in a Malian family
- Case Report-Gaucher Disease Type III C: Unusual cause of intracardiac calcification
- A Case of Type I Gaucher Disease Associated with Nonhodgkin Lymphoma
- Neonatal Gaucher Disease presenting as persistent Thrombocytopenia
- Bone Marrow Transplantation Corrected Gaucher Disease
Resources
[edit | edit source]
National Gaucher Foundation
2227 Idlewood Road, Suite 12
Tucker, GA 30084
[email protected]
Tel: 800-504-3189
Fax: 770-934-2911
National Organization for Rare Disorders (NORD)
P.O. Box 1968
(55 Kenosia Avenue)
Danbury, CT 06813-1968
[email protected]
Tel: 203-744-0100 Voice Mail 800-999-NORD (6673)
Fax: 203-798-2291
National Human Genome Research Institute – Learning About Gaucher Disease
1 http://www.genome.gov/25521505 1
National Institute of Neurological Disease and Stroke – Gaucher’s Disease Information
2 http://www.ninds.nih.gov/disorders/gauchers/gauchers.htm 2
- The National Institute of Neurological Disorders and Stroke (NINDS) support research innovative ways to treat and prevent lipid storage disorders. This includes clinical studies by the NINDS Developmental and Metabolic Neurology Branch.[2]
- Research on Gaucher disease and a link between Gaucher disease and Parkinson disease is currently being done at the Medical Genetics Branch of the National Human Genome Research Institute by Dr. Ellen Sidransky. Information about the research on Gaucher disease can be found at [Www.genome.gov/Staff/Sidransky www.genome.gov/Staff/Sidransky].[14]
References[edit | edit source]
- ↑ 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 National Human Genome Research Institute. Learning about gaucher disease. National Human Genome Research Institute: genome.gov. http://www.genome.gov/25521505. Updated September 2, 2010. Accessed March 5, 2011.
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 Office of Communications and Public Liaison. NINDS gaucher disease information page. National Institute of Neurological Disorders and Stroke: National Institutes of Health. http://www.ninds.nih.gov/disorders/gauchers/gauchers.htm. Updated October 27, 2010. Accessed March 2, 2011.
- ↑ Brave Community: Shire Human Genetic Therapies.Gaucher Disease Description. http://www.bravecommunity.com/condition/gaucher-disease/. Accessed April 1, 2011.
- ↑ 4.0 4.1 4.2 4.3 4.4 4.5 4.6 National Library of Medicine. Genetics home reference: Gaucher disease. National Library of Medicine: National Institutes of Health. http://ghr.nlm.nih.gov/condition/gaucher-disease. Updated January 2008. Published March 13, 2011. Accessed March 14, 2011.
- ↑ Genzyme Therapeutics Corporation. Gaucher disease information. fckLRhttp://www.cerezyme.com/healthcare/disease/cz_hc_disease.aspfckLRAccessed April 2, 2011.
- ↑ Kaneshiro, N.K., Zieve,D. Medline Plus: U.S. National Library of Medicine/National Institutes of Health: Gaucher Disease. http://www.nlm.nih.gov/medlineplus/ency/article/000564.htm. Updated November 12, 2010. Accessed April 1, 2011.
- ↑ 7.0 7.1 7.2 Genzyme Corporation. Gaucher disease diagnostic opportunities for the hematologist. http://www.makethediagnosis.com/hem/mtd_hem_opportunities.aspfckLR. Accessed April 2, 2011.
- ↑ http://jorieee.glogster.com/Gauchers-Disease-2/. Updated 2 years ago. Accessed April 1, 2011.
- ↑ Hughes, D. European Gaucher Leadership Forum: Unmet needs of the gaucher patient. Genzyme Corporation Europe. http://www.gaucherleadershipforum.org/healthcare/01/glf_hc_highlights01h.asp.fckLR Accessed April 4, 2011.
- ↑ 10.0 10.1 10.2 Cambridge University Hospitals: NHS Foundation Trust. Gaucher.fckLRhttp://www.cuh.org.uk/addenbrookes/services/clinical/lysosomal/disorders/gaucher.html. Accessed April 1, 2011.
- ↑ 11.0 11.1 11.2 11.3 11.4 11.5 Genzyme. Lysosomal Learning. fckLRhttp://www.lysosomallearning.com/healthcare/about/lsd_hc_abt_gaucher1.aspfckLRAccessed April 1, 2011.
- ↑ 12.0 12.1 14.Genzyme Corporation. Gaucher Care. fckLRhttp://www.gauchercare.com/en/healthcare/information/SignsAndSymptoms.aspx. Accessed April 1, 2011
- ↑ News Broadcast Network. Shire Announces FDA Approval of VPRIV for Gaucher Disease. http://www.youtube.com/watch?v=RVAX4_Q-woE. Accessed on April 2, 2011.
- ↑ 14.0 14.1 14.2 National Human Genome Research Institute. Learning about gaucher disease. National Human Genome Research Institute: genome.gov. http://www.genome.gov/25521505. Updated September 2, 2010. Accessed March 5, 2011.
- ↑ 15.0 15.1 15.2 15.3 15.4 15.5 15.6 15.7 15.8 15.9 Schmitz, J., Poll, W.L., Dahl, S. Therapy of adult Gaucher disease. Haematologica, Vol 92, Issue 2, 148-152 doi:10.3324/haematol.11193.fckLRhttp://www.haematologica.org/cgi/content/full/92/2/148/T10920148
- ↑ 16.0 16.1 Gansz, E.B. FDA Approves Therapy to Treat Gaucher Disease New drug will offer a treatment alternative for patients with rare genetic disorder. Food and Drug Admnistration. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm202288.htm. Updated April 9, 2010. Accessed April 1, 2011.
- ↑ 17.0 17.1 Pfizer, Inc. Treating gaucher disease. Pfizer, Inc: gaucherpartners.com. http://www.gaucherpartners.com/what-is-gaucher.aspx. Accessed March 5, 2011.
- ↑ Patterson, M.C. The Role of Small Molecule Inhibition in the Balance for Inborn Errors of Metabolism. Medscape Education. 2011. http://www.medscape.org/viewarticle/523700_2. Accessed April 2, 2011.
- ↑ 19.0 19.1 Accordant Health Services: CVS Caremark. Exercise and Gaucher Disease.fckLRhttp://www.cvscaremarkspecialtyrx.com/patients/condition-resources-tools/lysosomal-storage-disorders/gaucher-disease/staying-healthy/exerc. Updated April 30, 2010. Accessed April 1, 2011.
- ↑ 20.0 20.1 20.2 Pfizer, Inc. Gaucher Partners.com. Diagnosing Gaucher disease. http://www.gaucherpartners.com/diagnosing-gaucher.aspx. Accessed April 1, 2011.

